CE
Pharmacists: 1.00 contact hour (0.1 CEUs)
Released: May 22, 2025
Expiration: May 21, 2026
What Is Mastocytosis?
Jonathan A. Bernstein, MD:
Mastocytosis is a neoplastic disorder with substantial increase in and accumulation of clonal mast cells. When it only involves the skin, this disease is called cutaneous mastocytosis (CM). You may see other terms for CM, like urticaria pigmentosa, but the current recommendation is to use the term cutaneous mastocytosis when only the skin is involved.
When mystocytosis involves other extracutaneous organs, with or without skin involvement, we call it systemic mastocytosis (SM).
It is important to recognize that SM is not the same as mast cell activation syndrome. Mast cell activation syndrome is a hyperactivation disorder of clonal or nonclonal mast cells. Because symptoms are due to mast cell activation, they can be very similar to symptoms in SM, which is why it can be confusing for both patients and healthcare professionals (HCPs) to differentiate between these diseases.1,2
Classification of Systemic Mastocytosis
Jonathan A. Bernstein, MD:
This slide illustrates the classification of SM comparing the World Health Organization (WHO) with the International Consensus Classification (ICC) criteria.
Diagnoses of bone marrow mastocytosis, indolent SM (ISM), and smoldering SM are considered nonadvanced. The WHO classifies bone marrow mastocytosis as a distinct subtype, whereas the ICC includes bone marrow mastocytosis as a variant of ISM.
Advanced SM includes diagnoses of aggressive SM, which has more symptomatic presentation. Then there is SM with associated hematologic neoplasm (SM-AHN) and mast cell leukemia (MCL). These advanced versions are classified similarly by the WHO and ICC.3,4
Systemic Mastocytosis: Epidemiology and Pathway to Diagnosis
Jonathan A. Bernstein, MD:
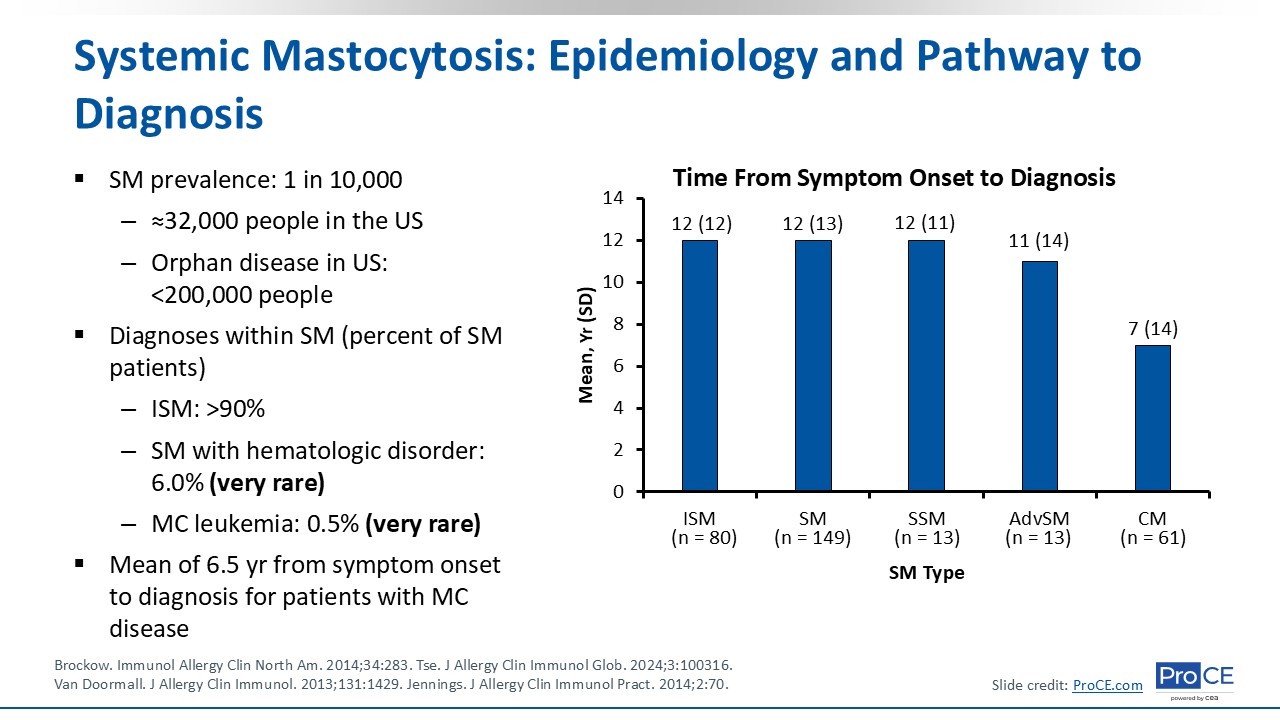
The prevalence of SM is approximately 1 in every 10,000 people, which translates to approximately 32,000 people in the United States. It is considered an orphan disease in the United States because it affects fewer than 200,000 people.
The most common diagnosis is ISM, which affects more than 90% of patients with SM.5 By contrast, SM with hematologic disorder occurs in approximately 6%, and MCL occurs in approximately 0.5% of patients with SM. Both are incredibly rare.
There is a mean of 6.5 years from symptom onset to diagnosis for patients with mast cell disease. This is largely because it presents with nonspecific symptoms and is not readily recognized by HCPs. The graph shows the mean time from symptom onset to diagnosis for the different variants of SM, and you can see that there is a delay in all SM subtypes.6,7
Why Is Systemic Mastocytosis Presentation So Heterogeneous?
Jonathan A. Bernstein, MD:
Multiple factors contribute to SM presentation heterogeneity. There is variability in mast cells and mast cell activation, matched with anatomic and microenvironmental impact on these cells. In addition, there are different surface receptors that direct mast cell response, including CD2, CD25, and CD30, and of course, there are different mediator profiles, too.
High mast cell counts in a range of tissues and organs may be associated with tissue and/or organ damage or failure caused by mast cell infiltration, and this is particularly evident in advanced forms of the disease.
Then there is mutational heterogeneity. Approximately 90% of patients with SM carry a somatic point mutation of KIT, which is the D816V variant. There also are somatic mutations in other genes that are associated with advanced SM and that could contribute to some of this variability.1,3,8
KIT Mutations as Drivers of Systemic Mastocytosis
Jonathan A. Bernstein, MD:
KIT mutations are drivers of SM. The KIT receptor is normally activated by stem-cell factor, leading to responses that result in mast cell activation, proliferation, survival, and migration to tissues. Mutations can lead to dysregulated, ligand-independent KIT receptor activation, with such activating mutations resulting in enhanced mast cell survival and accumulation.
It was mentioned before that D816V is the most common KIT somatic mutation in SM, and approximately 90% of patients with mastocytosis have this mutation. But there are others that HCPs also should consider in people who may have SM.3,9
Skin Lesions of Mastocytosis
Jonathan A. Bernstein, MD:
This slide shows skin lesions of mastocytosis, which is something that gets HCPs’ attention because patients often come into visits and ask, “I have all these freckles all over my body. What is going on here?”
These are maculopapular lesions. They are monomorphic and can be present as part of ISM or as isolated CM in adults. In general, maculopapular lesions are larger and more polymorphic in children.
I would say that more than 80% of patients with SM have cutaneous involvement, but some adults can present without any skin involvement, which makes this disease even more difficult to diagnose. In adults, the lesions often start on the thighs and spread to the trunk, distal extremities, and neck over time.10
Systemic Mastocytosis Clinical Presentation
Jonathan A. Bernstein, MD:
This table gives a nice overview of the clinical presentation of SM, including symptoms and associated mediators. For cutaneous symptoms, patients can present with a lot of itching and blushing, and they also may have hives or swelling, all of which involve histamine and tryptase. HCPs can measure 24-hour urine methylhistamine levels, 24-hour urine and serum tryptase levels, but other mediators related to mast cells are probably involved, including interleukin (IL)-6, platelet-activating factor (PAF), tumor necrosis factor (TNF), IL-8, IL-33, and prostaglandin D2 (PGD2).
Patients may present with different combinations of systemic symptoms, including syncope or near syncope, dizziness, lightheadedness, and/or racing heart rate. They may have gastrointestinal (GI) symptoms or musculoskeletal pain, and bone pain is also common. These patients often have osteopenia or osteoporosis, which can be accelerated on bone scans and dual-energy X-ray absorptiometry (DXA) scans. Patients frequently experience neurologic issues, which HCPs may dismiss as anxiety or depression instead of considering them as symptoms that are secondary to underlying SM. Patients can have respiratory symptoms, too.
When evaluating all of these symptoms and affected systems, they are not very specific, so HCPs must have a high degree of suspicion regarding SM. As shown in the table, there are overlapping effects of these different mediators that are released from mast cells that trigger many of these symptoms.11
Common Triggers in Systemic Mastocytosis
Jonathan A. Bernstein, MD:
Common triggers in mastocytosis include extremes or changes in temperature; stress; fatigue; certain foods and beverages; some medications; infection; hymenoptera venom, such as bee stings; and medical procedures, including surgery. With bee stings, or hymenoptera venom in general, HCPs should rule out underlying SM when patients present with anaphylaxis.7
Diagnostic Algorithm for Mastocytosis
Jonathan A. Bernstein, MD:
HCPs should have a high degree of suspicion for SM if they are seeing mast cell activation symptoms, which were described earlier, or anaphylaxis and/or elevated serum tryptase. Note that elevated serum tryptase is often seen but may not be present in all patients. Adult-onset mastocytosis in the skin should also increase suspicion for SM. If HCPs biopsy the lesions and identify positive increased numbers of mast cells, this should prompt for a more extensive evaluation. Although we can assess for an SM-related KIT mutation using a serum sample, the definitive test would be a bone marrow biopsy to determine if mast cells are increased.
Mast cell immunophenotyping and screening for FIP1L1::PDGFRA gene fusion are also recommended when evaluating a patient for SM. FIP1L1::PDGFRA screening is especially important if eosinophilia is present and test results for the KIT D816V mutation are negative.12
Diagnostic Criteria for Systemic Mastocytosis
Jonathan A. Bernstein, MD:
This slide outlines the diagnostic criteria for SM. The major criterion is the presence of multifocal dense mast-cell aggregates, with 15 or more mast cells per aggregate, in bone marrow or other extracutaneous organs. Note that mast-cell aggregates in the skin are excluded from this measure.
Minor criteria for SM include having 25% or more of one’s mast cells with atypical morphology; aberrant CD2, CD25, and/or CD30 expression on mast cells; presence of KIT D816V or other activating KIT mutation; and serum tryptase of 20 ng/mL or greater, if there is no myeloid neoplasm.
When looking at the WHO diagnostic criteria, patients must meet 1 major and 1 minor criteria or 3 or more minor criteria. This differs from the ICC criteria, which require patients to have only 1 major criterion or 3 or more minor criteria for diagnosis. I typically follow the WHO criteria for diagnosing SM.12
Identifying Systemic Mastocytosis Subtypes
Jonathan A. Bernstein, MD:
Here we have SM subtypes. With ISM, skin lesions are typical and there is usually at most 1 B-finding. In ISM without skin lesions, there is also a high basal tryptase and/or dense SM in an extramedullary organ.
For bone marrow mastocytosis, HCPs should not see skin lesions or B- or C-findings. This diagnosis really depends on patients’ basal tryptase, and diagnostic confirmation can be challenging if HCPs do not complete a bone marrow biopsy and have a high level of suspicion. In smoldering SM, patients typically have 2 or more B-findings but no C-findings.
SM-AHN typically is identified by bone marrow biopsy, and aggressive SM presents with 1 or more C-finding, with mast cells often in bone marrow smears. It is fortunate that SM-AHN and aggressive SM are rare.13